Researchers identify novel SARS-CoV-2 variant unregistered on genomic sequence databases
A team of scientists from the University of California Santa Cruz, USA, recently identified a novel variant (B.1.x) of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) that might be circulating in at least 20 US states and six countries globally. The mutations found in this variant are also present in other known variants of concern (VOCs). Crucially, because of the presence of a large deletion mutation, the sequence of B.1.x has been rejected by automated sequence checking tools used in publicly available genomic sequence databases. The study is currently available on the bioRxiv* preprint server.
-1.jpg)
Background
Whole genome sequencing of SARS-CoV-2 is one of the conventional methods to track viral evolution. A continuous sequencing of the viral genome is particularly important to identify the mutations that have emerged under positive selection and play vital roles in improving viral fitness, such as increased transmissibility and evasion of host immunity. In other words, early detection of new viral variants through genome sequencing is essential for understanding strain-specific clinical characteristics and developing strain-specific diagnostics and therapeutic and prophylactic interventions.
In the later phase of the coronavirus disease 2019 (COVID-19) pandemic, several new SARS-CoV-2 variants have been identified, with some showing significantly higher transmissibility and immune-evasion ability. These variants are designated as Variants of concern (VOCs) because of their severe impact on public health responses. The presence of multiple spike mutations is the most common feature among various VOCs, including the UK variant (lineage: B.1.1.7), the south African variant (lineage: B.1.351), and the Brazilian variant (lineage: P.1).
In the current report, scientists have presented the SARS-CoV-2 genome sequencing data obtained from an ongoing study.
Study design
The scientists analyzed high-quality SARS-CoV-2 sequences obtained from 339 samples from Santa Cruz County of California. About 58% of these sequences were from the B.1.427 and B.1.429 lineages, which were initially identified in California. Moreover, two of the tested sequences were associated with the B.1.1.7 lineage (the UK variant).
Important observations
By conducting genome sequencing, the scientists identified a novel variant of SARS-CoV-2 in eight samples. Because of its association with the B.1 lineage, they temporarily named the variant B.1.x. With further analysis, they observed that the novel variant shares several mutations with the UK variant and other known VOCs. Specifically, the variant exhibited several spike mutations, including S494P, N501Y, D614G, P681H, K854N, and E1111K. Of these mutations, N501Y is known to increase binding affinity between spike receptor-binding domain (RBD) and angiotensin-converting enzyme 2 (ACE2). Similarly, the D614G mutation, which was present in the globally dominant variant of SARS-CoV-2 in 2020, is known to increase viral infectivity. In addition, the B.1.x variant exhibited one nucleocapsid mutation (N:M234I), which is predicted to increase protein stability.
Regarding transmission dynamics of B.1.x, the scientists noticed a shape increase in frequency with time (<1% in January to >10% by mid-March). However, they failed to detect the variant in samples collected at the end of March. With further investigation, they observed that the increase in B.1.x frequency is primarily due to higher local transmission rather than multiple viral introduction events. Overall, these observations suggest that the number of cases with B.1.x may increase over time. By analyzing publicly available samples from different parts of the USA, they noticed that the variant is present in at least 20 states across the USA.
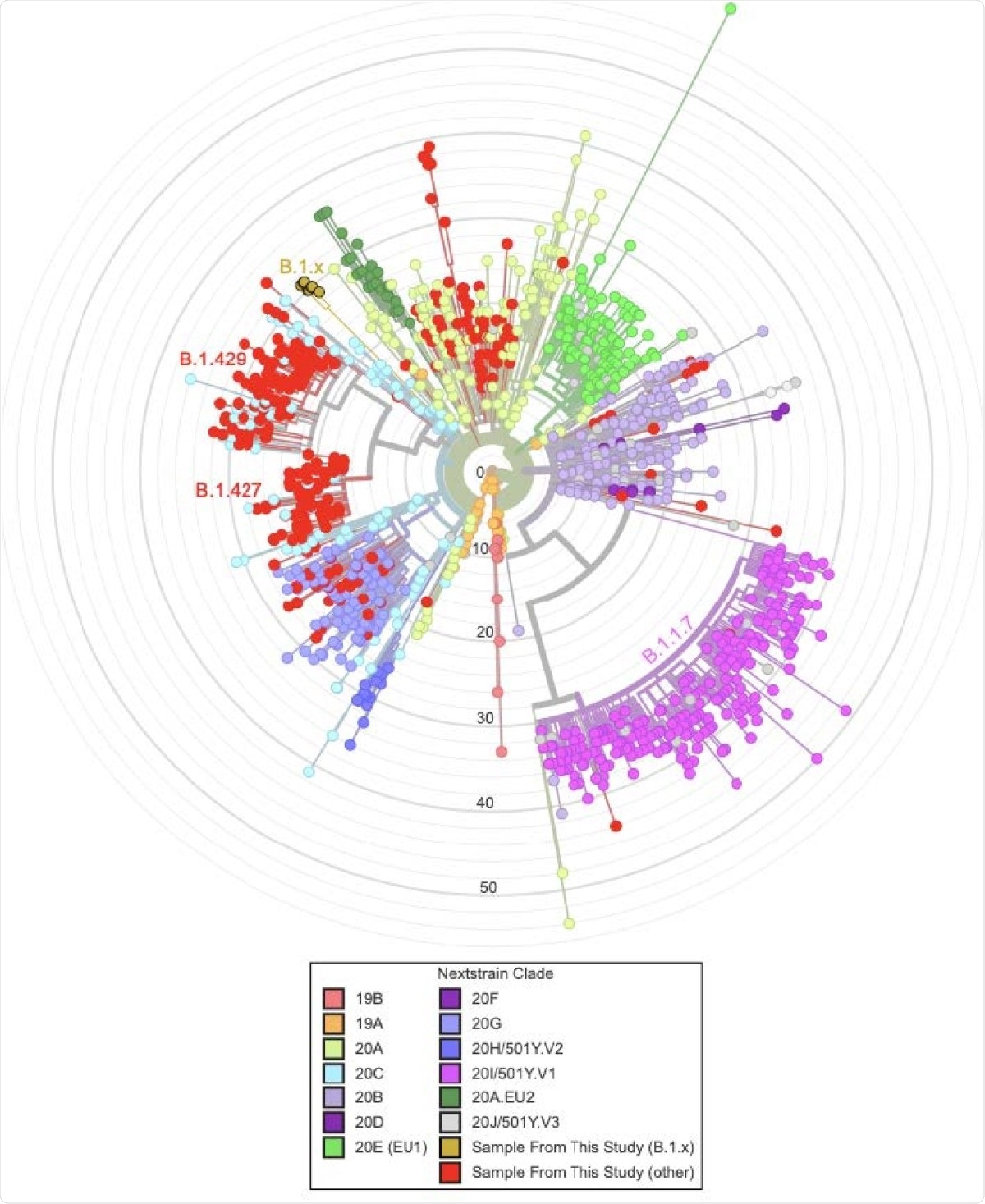
Most interestingly, the scientists observed that the variant contains a 35bp deletion causing a frameshift and a premature stop codon in the open reading frame 8 (ORF8). These characteristics are also present in the B.1.1.7 lineage because of a nonsense mutation. Such similarities between B.1.x and B.1.1.7 variants suggest that inactivation of ORF8 may be associated with viral evolution.
While submitting the sequences to the publicly available databases, the scientists noticed that both the GISAID and GenBank initially rejected all 8 genomes of the B.1.x variant because of the large deletion mutation in ORF8. This indicates that the sequences belonging to the B.1.x lineage are mostly underreported by these databases because of the technical problems associated with automated sequence quality control tools. To overcome such submission errors, the scientists suggest the use of UShER program that rapidly places new sequences onto an existing phylogeny. This will allow the corroboration between closely related sequences with novel mutations during batch submissions or individual sequence submissions.
Study significance
The study identifies a novel SARS-CoV-2 variant that shares genomic similarities with other known VOCs, such as the B.1.1.7. Based on the study findings, the scientists urge for rapid surveillance in order to understand the exact transmission dynamics and clinical implications associated with the novel SARS-CoV-2 variant.
*Important Notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Thornlow B. 2021. A new SARS-CoV-2 lineage that shares mutations with known Variants of Concern is rejected by automated sequence repository quality control. BioRxiv. doi: https://doi.org/10.1101/2021.04.05.438352, https://www.biorxiv.org/content/10.1101/2021.04.05.438352v1
Posted in: Medical Science News | Medical Research News | Disease/Infection News
Tags: ACE2, Angiotensin, Angiotensin-Converting Enzyme 2, binding affinity, Codon, Coronavirus, Coronavirus Disease COVID-19, Diagnostics, Enzyme, Evolution, Frequency, Genome, Genomic, Genomic Sequencing, Mutation, Pandemic, Phylogeny, Protein, Protein Stability, Public Health, Receptor, Respiratory, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Syndrome, Whole Genome Sequencing

Written by
Dr. Sanchari Sinha Dutta
Dr. Sanchari Sinha Dutta is a science communicator who believes in spreading the power of science in every corner of the world. She has a Bachelor of Science (B.Sc.) degree and a Master's of Science (M.Sc.) in biology and human physiology. Following her Master's degree, Sanchari went on to study a Ph.D. in human physiology. She has authored more than 10 original research articles, all of which have been published in world renowned international journals.
Source: Read Full Article